В современной офтальмологии для объяснения сложных
патогенетических механизмов развития различных глазных заболеваний (абиотрофий,
возрастных хориоретинальных дегенераций, воспалительных заболеваний
зрительного нерва и сосудистого тракта глаза, глаукомы и т.д.) все чаще
используется термин «апоптоз» – запрограммированная гибель клеток (от
греческого apoptosis –листопад)[Okisaka S, Murakami
A, Mizukawa A . Apoptosis in retinal
ganglion cell decrease in human glaucoma eyes. Jpn J Ophthalmol 1997 Vol
41:84-88]. Проблема исследования молекулярных механизмов
запрограммированной гибели клетки, а также взаимосвязь нарушения регуляции
этого процесса с различными заболеваниями (не только офтальмологическими)
волнует как специалистов в области молекулярной биологии, так и врачей всех
специальностей, в том числе и офтальмологов, что говорит о ее актуальности в
биологии и медицине.
Несмотря на большое количество экспериментальных данных, до
сих пор остаются неисследованными механизмы этого явления, не до конца выяснена
регуляция апоптоза отдельных клеток в целостном многоклеточном организме[Kerr F.R., Wyllie A.H., Currie A.R. Apoptosis: A basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer 1972; 26:239-256]. Кроме
того, до сих пор не определена позиция различных авторов в отношении самого
понятия «апоптоз». Является ли этот процесс «достойным» завершением жизни
клетки, когда её ресурсы исчерпаны, а задачи выполнены; или кроме этой
«благородной» смерти во имя жизни ещё существуют варианты, когда клетка,
казалось бы полная сил, погибает сама (пусть и без отягчающих обстоятельств -
некроза) и вовлекает в каскадный процесс массовых самоубийств своих, ни в чем
не повинных собратьев, которым бы жить и работать на благо целого организма?
Целью научной работы в этом направлении следует считать
изучение механизмов нарушения регуляции апоптоза, что позволит углубить
понимание этиологии и патогенеза ряда заболеваний для последующей возможной
коррекции этих нарушений и выработки патогенетически направленного лечения.
Апоптоз
- явление сложное и многогранное, возникшее в процессе эволюции с момента
появления многоклеточных организмов, служащее в целях регуляции естественного
баланса между размножением и гибелью клеток (необходимое условие поддержания
гомеостаза), а также для установления определенных взаимоотношений между
отдельными клетками в целостном организме [Агол В.И. Самопожертвование клетки
Химия и Жизнь №4 ,1997]. Именно благодаря этим взаимоотношениям клетки вступают
в различные этапы жизненного цикла - деления, роста, развития, дифференцировки,
старения и гибели. Старение в итоге приводит к катабиозу (греч.
"ката" - вниз, "биос" - жизнь) и смерти клеток [В. П.
Скулачёв Старение организма – особая биологическая функция, а не результат
поломки сложной биологической системы: биохимическое обоснование гипотезы
Вейсмана //Биохимия, 1997, том 62, вып. 11, с. 1394-1399
**. Хейфлик Л. Смертность и бессмертие на клеточном уровне // Биохимия, 1997,
том 62, вып. 11, с. 1380-1393,* Сеpов В. В. От целлюляpной патологии Виpхова
до молекуляpной патологии сегодняшнего дня АРХИВ ПАТОЛОГИИ №1 2001 стр.3-6].
Смертью клетки обычно называют необратимое прекращение явлений её жизнедеятельности.
Однако не всегда легко определить тот момент, когда клетка перестала жить. Даже
после растирания ткани до полного разрушения цитоплазматических мембран
(гомогенизации), метаболическая активность, проявляющаяся в поглощении
кислорода, брожении, гликолизе, ферментативной активности, не прекращается.
Надежным цитологическим критерием смерти клетки принято считать диффузную
окраску цитоплазмы и ядра витальными красителями (нейтральным красным,
метиленовым синим и т.п.). В живой же клетке эти красители накапливаются в
четко очерченных гранулах или вакуолях.
Следует заметить, что проблеме гибели клетки ученые уделяли
значительно меньше внимания, чем другим этапам её жизненного пути. Так, первые
высказывания о существовании процесса гибели клеток в многоклеточном организме
появились еще в конце XIX века. Однако годом признания апоптоза, как
физиологического явления, считается 1972 год, когда английские исследователи
Kerr, Wyllie, Currie [Kerr]
представили убедительные морфологические тому доказательства. Чуть ранее, в
1966 году, в журнале "Science" появилась статья J. Sanders
[Saunders, J.W. 1966. Death in Embryonic Systems. Science
154: 604-612.], в которой автор высказал предположение о том, что старение и
гибель клеток в эмбрио- и морфогенезе является результатом активации особой
генетической программы, запускаемой специфическими внутриклеточными и внешними
сигналами.
Во многих процессах онто- и эмбриогенеза апоптоз играет
важнейшую роль для поддержания гомеостаза. Апоптотическую гибель клеток можно
проследить в эмбриональном развитии у беспозвоночных животных. Изучена также
запрограммированная клеточная гибель в процессе эмбриогенеза высших позвоночных
- при развитии глаза, сердца и нервной системы млекопитающих. Нарушение
процесса апоптоза в эмбриогенезе может привести к внутриутробной гибели плода,
врожденным уродствам, а также к различным заболеваниям, в том числе и
злокачественным новообразованиям [Хейфлик Л. В кн. Молекулы и клетки, Вып. 7,
М., "Мир", 1982, 134-148.].
Во
взрослом организме физиологический механизм апоптоза можно проследить в
различных типах тканей, как в медленно пролиферирующей популяции клеток
(гепатоциты, клетки эпителия коры надпочечников), так и в быстро
пролиферирующих клеточных популяциях. В первом случае он выполняет функцию
гомеостатической регуляции оптимального объема ткани. Во втором роль апоптоза,
в основном, связана с дифференцировкой клеток [Белушкина Н.Н Апоптоз в
многоклеточном организме//Архив патологии, №1, т.63, стр.51-60]. Путем
программирования клеточной гибели происходит удаление клеток, выживание которых
нежелательно для организма, например мутантных клеток или клеток, зараженных
вирусом. В последнем случае этот процесс имеет важное биологическое значение,
поскольку фрагментация пораженной ДНК во время процесса апоптотической гибели
предупреждает перенос генетического материала в другие клетки.
Роль апоптоза можно проследить и в
патогенезе различных заболеваний (явление апоптотической гибели клеток
наблюдается не только при физиологических, но и при различных патологических
состояниях). Гибель клеток путем апоптоза происходит при уменьшении
кровоснабжения органа, в том числе при различных ишемических состояниях глаза
(тромбозы ЦВС и окклюзии ЦАС и их ветвей; хронические и острые нарушения
кровообращения в зрительном нерве и т.д.), при глаукоме, абиотрофиях,
катаракте, ретинобластоме, диабетической ретинопатии [Nickells
R.W.; Zack
D.J. Apoptosis
in ocular
disease: a
molecular overview. Ophthalmic-Genet.
1996 Dec; 17(4): 145-65, Levin L.A.; Louhab A. Apoptosis of retinal ganglion
cells in anterior ischemic optic neuropathy. Arch-Ophthalmol. 1996 Apr; 114(4):
488-91]. Механизм запрограммированной
гибели клеток прослеживается и в эндокринно-зависимых тканях (органах- мишенях)
при уменьшении концентрации соответствующего гормона (например, клеток простаты
после кастрации и коры надпочечников после подавления синтеза АКТГ
глюкокортикоидами). Воздействие радиации вызывает апоптоз в пролиферирующих
клетках эпителия кишечника и в непролиферирующих клетках иммунной системы.
Можно заключить, что апоптоз является
широко распространенным общебиологическим механизмом, ответственным не только
за поддержание постоянства численности клеток, формообразование, выбраковку
дефектных клеток, но и за развитие различных патологических состояний отдельных
клеток, систем и организма в целом [Narayanan V.
Apoptosis in development of the nervous system. 1. Naturally occurring
death in the developing nervous system. Pediatr Neurol 1997; Vol 16 P.
9-13.]
Согласно
данным морфологического анализа установлены две основные причины клеточной
гибели: некроз и апоптоз. При некротической форме гибели клетки набухают.
Вследствие нарушения работы ионных каналов митохондрии и другие органеллы
расширяются, что приводит к разрыву внутриклеточных механизмов и плазматической
мембраны клетки. Активируются лизосомальные ферменты. Это приводит к тому, что
внутриклеточное содержимое, попадая во внеклеточную среду, способствует
развитию воспаления. Причины, приводящие к некрозу клетки: гипертермия;
ингибирование окислительного фосфорилирования, гликолиза или цикла Кребса;
гипоксия; действие комплемента или различных токсинов и другие. [Белушкина;
Гусев-Скворцова].
Отличительным морфологическим проявлением апоптоза является
коллапс ядра. После получения сигнала, призывающего клетку подвергнуться
апоптозу, в ней происходит целый ряд биохимических и морфологических изменений.
В отличие от некроза, при апоптозе клетка сморщивается, теряя в течение
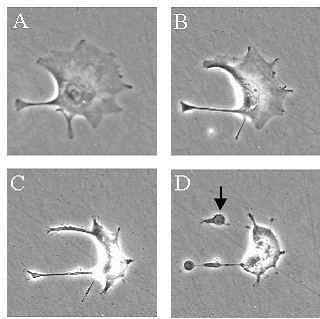
нескольких минут до 1/3 своего объема ( рис.1, А). “Усыхание” хорошо выражено
как в культуре клеток, так и в тканевых срезах, где апоптотическая клетка
отделяется от соседних клеток. Хроматин, который в норме представлен открытыми
и конденсированными областями (гетеро- и эухроматин), становится
суперконденсированным, принимает форму полумесяца (подковы) по периферии ядра
(рис.1, В).
Рисунок 1. Этапы апоптоза
трофобластической клетки. Стрелкой обозначено апоптотическое тельце. (Фото выполнено Nitric Oxide Research Group, St George’s hospital medical Scool,
University of London)
Вследствие
активации семейства белков, именуемых каспазами (известно около 10 видов
каспаз) начинаются процессы протеолиза, приводящие к гидролизу структурных
белков клетки. Кульминацией можно считать активизацию нуклеаз, когда
начинается фрагментация геномной ДНК, что является биохимическим маркером
апоптоза [Negoescu A., Guillermet C., Lorimier P. Importance of DNA fragmentation in apoptosis with regard to TUNEL specificity. Biomed-Pharmacother, 1998,
Vol.52, №6, p.252-258]. Это явление необратимо, оно
продолжается до наступления изменений в клеточной проницаемости, что и приводит
клетку к гибели. В большинстве клеток такую фрагментацию ДНК вызывают кальций и
магний-зависимые ядерные эндонуклеазы, которые избирательно «разрезают» ДНК на
отрезки, локализованные между нуклеосомами (линкерные участки ДНК), и приводят
к образованию моно- и олигонуклеосомных фрагментов ДНК
[Steller, H. 1995. Mechanisms and genes of cellular suicide. Science
267: 1445-1449]. Процесс активации ферментов тонко регулируется и является
АТФ-зависимым. Нуклеазной атаке подвергаются не только эухроматиновые
(генетически активные), но и спирализованные уплотненные гетерохроматиновые
участки. Для того, чтобы запустить этот процесс, клетка должна произвести
ферменты – нуклеазы, что приводит к усилению процессов транскрипции (биосинтез
РНК) и трансляции (биосинтез белка). Имеются данные, что ингибиторы белкового
синтеза - циклогексамид и пуромицин - предотвращают энзиматический распад
хроматина и могут предотвратить или отсрочить процесс апоптоза. Хлористый цинк
также приводит к предотвращению фрагментации ДНК и отсрочивает апоптоз
[Животовский Б.Д., Хансон К.П. Сб. "Биополимеры и клетка", 1, 1985
№4, 199-203;].
Программируемая клеточная смерть может иногда
приостанавливаться при ингибировании транскрипции или трансляции нуклеиновых
кислот [Steller, H. 1995. Mechanisms and genes of cellular suicide. Science 267: 1445-1449]. Это
свидетельствует о том, что гибель клетки регулируется клеточными механизмами.
Клетки продолжают сокращаться (рис.1, C), принимая форму,
удобную для фагоцитирования их макрофагами (или соседними клетками). Последние,
в свою очередь, ответственны за удаление апоптотических клеток из тканей крайне
«деликатным» способом. Изменения на уровне мембран клеток могут наблюдаться
морфологически в виде появления специфических мембранных выпячиваний (D) или
пузырей, которые часто завершают апоптотический процесс. Небольшие пузырьки
часто называют «апоптозными тельцами» ( стрелка, D) [Lash, G. E., J. E. Cartwright, et al. (1999). “The effects of angiogenic growth factors on extravillous
trophoblast invasion and motility.” Placenta 20(8): 661-667]. На данном этапе клетка еще жива (включение
летального красителя трипанового синего не происходит). Далее еще живые
апоптозные тельца утилизируются, содержимое клетки не попадает во внеклеточную
среду и не вызывает воспалительных явлений. Таким образом, уничтожение клеток
путем апоптоза обеспечивает минимальное повреждение тканей и является более
«деликатным» по сравнению с некротическим механизмом гибели.
Исследования продемонстрировали, что процесс апоптоза
генетически детерминирован и регулируется следующими генами: усиление работы
генов Cip1 (p21), Bax (p21), Daax, FAF-1, FADD, TRADD, RAIDD, RIP, SIVA, FLIP,
CAS, TIA-1/TIAR, TDAG51 и др., белков p53, р21 ускоряет течение апоптоза, гены
семейства Bcl-2, Bad, Bag1 препятствуют апоптозу [Macaya A. Apoptosis in the nervous system Rev-Neurol.
1996 Nov; N24
Vol 135 P. 1356-60].
Важен
и тот факт, что после начального повреждения нередко включается некий
триггерный механизм. Он приводит к тому, что каскадная гибель клеток может
продолжаться даже после удаления первичного стимула нейронного апоптоза [Schwartz M., Belkin M., Yoles E. Potential treatment modalities for glaucomatous neuropathy: neuroprotection and neuroregeneration. J Glaucoma 1996;5:427-432.]. При
исследовании патологических процессов, происходящих в нейронах центральной
нервной системы, выявлены механизмы, действующие в случае так называемой
вторичной дегенерации, когда изначально локальное повреждение одних нейронов
распространяется на смежные интактные (первично неповрежденные) нейроны.
Возможно, этим механизмом объясняется прогрессирующее снижение зрительных
функций у пациентов с глаукомой, компенсация внутриглазного давления у которых
была достигнута тем или иным способом [Brubaker RF. Delayed
functional loss in glaucoma. LII Edward Jackson Memorial Lecture. Am J
Ophthalmol 1996;121: 473-483].
Мнения исследователей по вопросу о факторах, индуцирующих
апоптоз, представлены в литературе достаточно широко. Это могут быть
неспецифические факторы, такие как температура, токсические агенты, оксиданты,
свободные радикалы, гамма-, рентген- и УФ-излучение[Macaya A. Apoptosis in the nervous system Rev-Neurol.
1996 Nov; N24 Vol
135 P. 1356-60], бактериальные токсины и др. Во всех
этих случаях происходит индукция апоптоза, но при увеличении дозы
соответствующего агента может развиться некроз. "Неспецифичность"
этих воздействий заключается в том, что они не имеют адресной мишени в клетке и
индуцируют множественные поражения генома, белков или (и) вызывают
биоэнергетическую катастрофу. Поскольку апоптоз - физиологическое явление, то в
организме имеются и специфические факторы, приводящие к апоптозу клетки. К
настоящему времени известно, что апоптоз могут вызывать как внутриклеточные
сигналы, так и внешние, сами по себе не являющиеся токсическими или
деструктивными, и опосредующие свое действие через рецепторные системы. В
целом, если идет речь о физиологических факторах, то, вероятно, следует
применять термин регуляция апоптоза клетки, поскольку известна группа
физиологических активаторов и ингибиторов апоптоза.
В литературе приводятся некоторые факторы
индукции апоптоза при глаукоме. Повышение внутриглазного давления провоцирует
апоптоз: прямая компрессия аксонов ганглиозных клеток искривленными ламинарными
перегородками вследствие повышенного внутриглазного давления не только снижает
аксоплазматический ток [Minckler D.S. Bunt A.H. Orthograde and retrograde axoplasmatic transport during acute ocular hypertension in the monkey. Invest Ophthalm Vis Sci 1977 Vol.16, P.426], но и вызывает снижение ретроградного аксонального
транспорта. Это, в свою очередь, приводит к уменьшению доставки к телу
ганглиозной клетки сетчатки нейротрофических факторов. Недостаток последних
включает пусковой механизм апоптоза и приводит к программной гибели клеток [Garcia-Valenzuela E.; Shareef S.; Walsh J. Programmed cell death of retinal ganglion cells during
experimental glaucoma. Exp-Eye-Res. 1995 Jul; 61(1): 33-44]. Апоптоз в
этом случае идентифицирован по следующим специфическим признакам: конденсации
хроматина, дроблению геномной ДНК, обнаружению апоптозных телец [Quigley H.A. Ganglion cell death in glaucoma: pathology recapitulates
ontogeny Aust-N-Z-J-Ophthalmol. 1995 May; 23(2): 85-91, Importance
of DNA fragmentation in apoptosis with regard to TUNEL specificity * Negoescu
A.; Guillermet C.; Lorimier P.; Biomed-Pharmacother. 1998; Vol 52 N6 P. 252-8].
Имеются данные о том, что
нейротрофические факторы, тормозящие апоптоз в нейронах взрослого организма,
оказывают большое влияние на развитие сетчатки и в период эмбриогенеза: из-за
неадекватной трофической стимуляции происходит нейрональный апоптоз и в
развивающихся ганглиозных клетках [Frade JM, Bololenta P, Martinez-Morales JR, et
al. Control of early cell death
by BDNF in chick retina. Development
1997;124:3313-3320]. Этот факт еще раз подтверждает наличие сложных
взаимозависимых связей в системе «глаз – мозг». Об «…особой, ключевой роли
блокирования аксоплазматического (орто-, и ретроградного) тока в волокнах
зрительного нерва» у больных глаукомой пишет В.В. Волков [В.В. Волков Глаукома
при псевдонормальном давлении. М., Медицина, 2001, 350стр.]. Таким образом, при
нарушении ретроградного аксонального тока ганглиозная клетка сетчатки не только
не получает информацию о дистальных отделах аксона [Miller N.R.//Walsh and Hoyts clinical neuroophthalmology. 1982, Vol.1, P.175-176], но и лишается
жизненно важной поддержки нейропротекторными факторами, блокирующими апоптоз.
Нейропротекторные факторы, которые стимулируют выживание ганглиозных клеток
сетчатки человека в культурах ткани, включают основной фактор роста
фибробластов (bFGF), производимый мозгом нейротрофический фактор (BDNF) и цилиарный нейротрофический фактор. Сколько и какие именно трофические факторы происходят из
латерального коленчатого тела (ЛКТ) и транспортируются назад, к сетчатке,
неизвестно. Однако in vitro нейротрофический фактор мозга оказался наиболее
действенным при защите ганглиозных клеток сетчатки от смерти после перерезки их
аксонов [Quigley H.A.; Nickells R.W.; Kerrigan L.A Retinal ganglion cell death in experimental glaucoma and
after axotomy occurs by apoptosis. . Invest-Ophthalmol-Vis-Sci. 1995 Apr; 36(5): 774-86]. Имеются данные, что при эмбриональном развитии сетчатки
выживают те ганглиозные клетки, растущие аксоны которых контактируют с ЛКТ,
где, вероятно, терминальные участки аксонов ганглиозных клеток сетчатки и
подвергаются стимуляции нейротрофическим фактором мозга. Ганглиозные клетки сетчатки, которые не получили стимуляции вышеназванным трофическим фактором, подвергаются апоптозу [Rait
J. The prevention of ganglion cell suicide in primary open
angle glaucoma Asian J
Ophthalmol 1998;1(2):3-8 Pienardo-Ramon R, Salvador
M., Villegas-Perez MP Effects of axotomy and intraocular administration of
NT-4, NT-3 and BDNF on the survival of adult rat retinal ganglion cells: A
quantitative in-vivo study. Inv Ophthalmol Vis Sci 1996; 37:489-500].
Предполагается, что наличие такой
трофической поддержки является необходимым условием для выживания ганглиозных
клеток сетчатки, а нарушения аксоплазматического транспорта могут стимулировать
апоптоз в восприимчивых к дефициту трофической стимуляции клетках [Pienardo-Ramon R, Salvador M,
.]. Возникает справедливый вопрос: отличается ли толерантность ганглиозных
клеток сетчатки различных особей к дефициту трофической стимуляции? Если да, то
чем это предопределяется? К сожалению, ответа на этот вопрос авторы статьи в
литературе не нашли.
Некоторые исследователи связывают неадекватную
нейротрофическую стимуляцию с митохондриальной дисфункцией и, вследствие этого,
появлением внутриклеточного энергетического дефицита [Wadia J.S.,
Chalmers-Redman R., Ju W., et al. Mitochondrial membrane
potential and nuclear changes in apoptosis caused by serum and nerve growth
factor withdrawal: time course and modification by (-) -Deprenyl. J Neurosci
1998;18:932-947.]. В клетках иных (не нервной) тканей снижение
митохондриального мембранного потенциала является одним из самых ранних
признаков начала апоптоза. Снижение производства энергии вызывает повышение активности кислорода и может стать пусковым механизмом развития апоптоза как в
здоровых, так и в больных клетках [Schapira,-A-H Mitochondrial disorders. Curr-Opin-Neurol. 1997 Feb; 10(1): 43-7 ].
Среди растущего числа выявленных пусковых механизмов апоптоза
выделяются некоторые факторы, играющие в этом процессе ведущую роль. Выделены
аминокислоты (в генезе глаукомы имеет значение глутаминовая кислота или глутамат),
избыток которых патологически повышает деполяризацию клеточной мембраны. Кроме
того, в больших количествах они являются токсинами, что дает возможность
признать их роль в происхождении многих неврологических нарушений. Повышение
концентрации этих аминокислот приводит к накоплению калия, кальция и
освобождению свободных радикалов внутри клеток. При первичной открытоугольной
глаукоме «глутаматовая токсичность» играет основную роль в том «каскаде»
вторичной дегенерации ганглиозных клеток сетчатки, о котором говорилось выше.
Следует заметить, что глутамат является универсальным нейромедиатором нервной
системы. В норме он является трансмиттером при передаче возбуждения от
зрительных рецепторов к биполярам и горизонтальным клеткам. Глутамат используется
также и биполярами при контакте с амакриновыми клетками сетчатки. Ганглиозные
клетки сетчатки используют иные нейромедиаторы: g-аминомасляную кислоту
(ГАМК), глицин, допамин [Sieving P. A. Electrical Signals of the Retina and Visual Cortex. Digital Duane’s Ophthalmology Vol. 2, Chapter 13]. Не ясно, почему глутамат в повышенных концентрациях наиболее
токсичен для ганглиозных клеток сетчатки; можно лишь предположить, что
последние наименее адаптированы к его присутствию. Хотя могут существовать и
другие объяснения факту индукции апоптоза ганглиозных клеток сетчатки при
повышенной концентрации глутамата.
Dreyer [Dreyer EB, Zurakowski D, Schumer
RA, et al. Elevated glutamate levels in the vitreous body of humans and monkeys
with glaucoma. Arch Ophthalmol 1996; 114:299-305.] в 1996 году одним из первых
сообщил о повышении концентрации глутамата в стекловидном теле глаз больных
ПОУГ, обнаруженном автором при экстракции катаракты. В этой же статье Dreyer отмечает, что в заднем
отделе стекловидного тела обезьян с нелеченной (моделированной в экперименте)
глаукомой было также выявлено 7-ми кратное превышение концентрации глутамата по
сравнению с нормой, что вызывало токсическое повреждение ретинальных
ганглиозных клеток. Активация глутаматом N-Methyl-D-Aspartate (NMDA) рецепторов увеличивает концентрацию внутриклеточного
кальция и калия, приводит к повышению концентрации свободных радикалов, что, в
свою очередь, включает механизм апоптотических ядерных превращений. К
сожалению, лекарственные средства, которые длительно или необратимо блокируют NMDA-рецепторы, также вызывают серьезные побочные эффекты: нейротоксикоз,
дисфункции нервных элементов, психозы или летальный исход.
Остается открытым вопрос: почему в стекловидном
теле больных ПОУГ повышается концентрация глутамата? Что первично: увеличение
количества глутамата (механизм этого увеличения?) и его роль как нейротоксина -
инициатора апоптоза; либо концентрация этого вещества вторично возрастает при
каскадном характере гибели ганглиозных клеток сетчатки, что ещё более
потенциирует апоптоз?
К физиологическим факторам, способным
запускать в клетках апоптотическую программу, относится также оксид азота (NO)
- одна из ключевых сигнальных молекул, регулирующих функции сердечно-
сосудистой, нервной и иммунной систем организма. Оксид азота участвует в
нейротрансмиссии в центральной и периферической нервных системах [Snyder,
Solomon H., "Nitric Oxide: First in a New Class of
Neurotransmitters?", Science, Vol. 257, 1992, P.
494-496. ** Ott, S.R., Jones, I.W.,
Burrows, M. and Elphick, M.R. () Sensory afferents and motor neurons as targets
for nitric oxide in the locust. J. Comp. Neurol. 2000 Vol. 422, P.521-532.].
S. Snyder, директор института нейробиологии J.
Hopkins Medical School отметил: «За мои 25 лет
работы в нейробиологии я не знал другую молекулу, которая так активно влияла бы
на физиологические и патологические функции организма». В 1992 году журналом Science NO
была признана «Молекулой Года». Уникальная химическая природа и большое число
внутриклеточных мишеней для NO, а также её физиологически активных форм,
оставляют открытым вопрос, каким образом и сколь специфичным оказывается
повреждающее действие оксида азота на клетки. Первоначально
вещество, способствующее вазодилятации и регулирующее сосудистый тонус, было
названо эндотелиальным фактором дилятации (EDRF),
считаясь белком. Впоследствии выяснилось, что это EDRF - оксид азота, довольно нестойкая молекула газа с периодом полураспада в
нескольких секунд. Перенос ионов кальция внутрь клетки повышает синтез оксида
азота из аргинина при участии фермента- NO-синтазы (NOS)[].
В силу своих небольших размеров NO- молекула липофильна, способна проникать через
мембраны клетки и реагировать с множеством биохимических мишеней, в том числе с
гемогруппами, железными и цинковыми кластерами. Нарушение синтеза или регуляции
количества NO способно воздействовать на множество важных биологических
процессов и приводить к различным заболеваниям [Реутов В.П. Цикл окиси азота в
организме млекопитающих // Усп. биол. наук. —1995 .— Т. 35. —С.
189—228.* Baum R. Superoxide theory of oxygen toxicity is center of heated
debate. // Chem. Engin. News.1984. V. 9. P. 20-28*.Lewen
A.; Matz P.; Chan P. J-Neurotrauma. 2000 17(10) P.871-90].Доказана роль оксида азота в
регулировании нейроапоптоза. При этом в зависимости от концентрации NO и типа
клетки, оксид азота может как стимулировать апоптоз, так защищать клетку от
смерти [Neufeld A.H. Nitric
oxide: a potential
mediator of
retinal ganglion
cell damage
in glaucoma. Surv-Ophthalmol.
1999 Jun; 43 Suppl 1S129-35]. Блокада
NO-синтазы ингибирует апоптоз,
в то же время усиливает действие нейротрофических факторов
(BDNF) на ганглиозные клетки сетчатки
[Klocker N, Cellerino A, Bahr M. Free radical
scavenging and inhibition of nitric oxide synthase potentiates the
neuro-trophic effects of brain-derived neuro-trophic factor on axotomized
retinal ganglion cells in vivo. J Neurosci 1998;18:1038-1046.]. Выявлено, что окись азота способна также ингибировать
апоптоз различных типов клеток: лейкоцитов, гепатоцитов, эндотелиальных
клеток. Антиапоптотические эффекты NO могут
быть опосредованы через множество механизмов, например, путем инактивации
многих из белков - каспаз, включая каспазу 3, каспазу 1 и каспазу 8. В
больших концентрациях NO-
мощный нейротоксин. Кроме того, оксид азота при определенных концентрациях
снижает тонус сосудов глаза, уменьшая перфузию, изменяет сопротивление оттоку
камерной влаги, сокращает плотность ганглиозных клеток сетчатки (эти эффекты
крайне нежелательны при глаукоме) [Волков В.В. Глаукома при псевдонормальном
давлении. Руководство для врачей. Москва, М., 2001, 350 стр.].
Среди прочих факторов индукции апоптоза следует отметить
увеличение концентрации продуктов перекисного окисления липидов (ПОЛ) и
свободных радикалов - активных форм кислорода (АФК) в водянистой влаге передней
камеры глаза [Бунин А.Я. Метаболические факторы патогенеза первичной
открытоугольной глаукомы // Глаукома Материалы Всероссийск. Науч.-практич.
Конференции «глаукома на рубеже тысячелетий; итоги и перспективы. 1999,
стр.9-12]. ПОЛ – необходимый процесс поддержания гомеостаза: при обновлении
липидного состава клеточных мембран сохраняется их нормальная проницаемость и
активность мембранно-связанных ферментов. При различной патологии интенсивность
ПОЛ резко возрастает вследствие увеличения концентрации активных форм
кислорода.
Повреждающими же агентами являются активные формы кислорода
(АФК), образующиеся в ряде физико-химических процессов в организме. Главные
активные формы кислорода [Осипов А.Н., Азизова О.А., Владимиров Ю.В.
Активные формы кислорода и их роль в организме.//Успехи.биол.химии. 1990. Т.
31. С. 180-208]:
- гидроксильные
(свободные) радикалы (ОН-),
- супероксидные
радикалы (О2-),
- перекись
водорода (Н2О2),
- синглетные
формы кислорода (1О2),
- пергидроксильные
ионы (НО2-).
Основные механизмы появления АФК в организме обычно связаны с
нарушениями функционирования электронно-транспортных цепей митохондрий или
микросом, а также с изменением свойств дегидрогеназ.
Синглетный кислород образуется в реакциях фотоокисления в присутствии так
называемых фотосенсибилизаторов: флавинов, гематопорфиринов и др., а также при
дисмутации супероксидных радикалов [Khan A.U. //Science.1970. Vol.168.
P.476-482. 1970]. Синглетный кислород агрессивен в отношении биосубстратов, в
особенности в отношении молекул с двойной связью. Конечным итогом таких реакций
обычно является образование гидроперекисей органических молекул – одного из
важнейших моментов в процессах перекисного окисления ненасыщенных липидов в
биомембранах [Осипов,1990; Roschupkin D.I., Pelenitsin A.B., Potapenko A. //
Photochem.and Photobiol. 1975. Vol.21. P.63-69]. В присутствии металлов
переменной валентности АФК запускают цепные реакции окислительной деградации
биомолекул [Владимиров Ю.А., Арчаков А.И. Перекисное окисление липидов в
биомембранах. М.: Наука. 1972].
Во всех клетках с аэробным типом метаболизма в процессе присоединения
одного электрона к молекуле кислорода образуются супероксидный анион-радикал -
О2- и его протонированная форма – гидроперекисный радикал
- НО2-. Оба они порождают ряд других активных форм
кислорода. Образование этих АФК наиболее существенно вблизи цепей переноса
электронов – дыхательная цепь, микросомы [Каган В.Е., Сербинова А.Е.,Минин А.А.
и др. //Биохимия. 1985. Т. 50. С. 986-991. Rosen G.M., Finkelstein E., Rauckman E.J.//Arch.Biochem.
and Biophys. 1982. Vol.215. P.367-379.]. Несмотря на то, что эти АФК
играют важную роль в неспецифических иммунных механизмах организма,
они являются весьма активными соединениями и обладают высокой биологической
агрессивностью. Высокая реакционная способность гидроксильного радикала (ОН-),
образующегося из перекиси водорода в присутствии ионов металлов, вызывает
прямое повреждение ДНК с разрывом цепи и лецитина – главного компонента
биологических мембран. С другими биомолекулами (ОН-) образует
вторичные свободные радикалы, в том числе перекисные соединения липидов
[Азизова О.А.,Осипов А.Н., Савов В.М. и др. // Биофизика. 1985. Т. 30.
С.36-39].
В связи с особой реакционной способностью АФК, их повышенное содержание
во влаге передней камеры является не только патогенетическим фактором
катарактогенеза, но и первичной открытоугольной глаукомы [Бабижаев М.А. //
Вопросы мед. химии- 1985.-№1.- стр.15]. При этом активные свободные радикалы,
оказывая цитотоксическое действие на эндотелий, покрывающий коллагеновые
волокна трабекулярного аппарата, индуцируют его (эндотелия) апоптотическую
гибель. Некоторые авторы отмечают нарастание концентрации продуктов ПОЛ по мере
прогрессирования глаукомы [Бунин А.Я., Бабижаев М.А, Супрун А.В.//
Вестн.офтальм. 1985 №2 Стр. 13- 16]. Нельзя забывать о триггерном механизме
апоптоза, когда за гибелью отдельных клеток наступает «каскад» самоуничтожения
здоровых клеток. Этот факт может открыть дискуссию на тему о сроках оперативного
лечения глаукомы и явиться весомым аргументом не в пользу хирургии на поздних
стадиях болезни.
Естественно, в статье приведены не все факторы, прямо или косвенно
приводящие к программной гибели клеток. Некоторые из них очень важны и
бесспорны, как, например, митохондриальные дисфункции (некоторые
антиапоптотические факторы - Bcl-XL и
др.- расположены на внешней стороне мембраны митохондрий). Другие факторы
достойны внимания, но спорны (интенсивный прерывистый световой поток, по
данным Li S.,
Chang C.
[Li S.;
Chang C.J.; Abler A.S.; Fu J. A comparison of continuous versus intermittent light exposure on apoptosis Curr-Eye-Res. 1996 Sep; 15(9): 914-22] также
стимулирует триггерный апоптоз фоторецепторов сетчатки). Тем не менее, новые
факты дополняют метаболическую теорию патогенеза глаукомы, что открывает
широкие возможности для лечения этого заболевания.
1.
Агол В.И.
Самопожертвование клетки Химия и Жизнь №4 ,1997
2.
Азизова
О.А.,Осипов А.Н., Савов В.М. и др. // Биофизика. 1985. Т. 30. С.36-39
3.
Бабижаев М.А. //
Вопросы мед. химии- 1985.-№1.- стр.15
4.
Белушкина Н.Н
Апоптоз в многоклеточном организме//Архив патологии, №1, т.63, стр.51-60
5.
Бунин А.Я.
Метаболические факторы патогенеза первичной открытоугольной глаукомы //
Глаукома Материалы Всероссийск. Науч.-практич. Конференции «глаукома на рубеже
тысячелетий; итоги и перспективы. 1999, стр.9-12
6.
Владимиров Ю.А.,
Арчаков А.И. Перекисное окисление липидов в биомембранах. М.: Наука. 1972
7.
Волков В.В. :
Глаукома при псевдонормальном давлении. Руководство для врачей. Москва, М.,
2001, 350 стр.
8.
Гусев Е.И.,
Скворцова В.И. : Ишемия головного мозга. Москва, М., 2001. 326 стр.
9.
Животовский Б.Д.,
Хансон К.П. Сб. "Биополимеры и клетка", 1, 1985 №4, 199-203
10.
Каган В.Е.,
Сербинова А.Е.,Минин А.А. и др. //Биохимия. 1985. Т. 50. С. 986-991. Rosen G.M.,
Finkelstein E., Rauckman E.J.//Arch.Biochem. and Biophys. 1982. Vol.215. P.367-379
11.
Осипов А.Н.,
Азизова О.А., Владимиров Ю.В. Активные формы кислорода и их роль в
организме.//Успехи.биол.химии. 1990. Т. 31. С. 180-208
12.
Реутов В.П. Цикл
окиси азота в организме млекопитающих // Усп. биол. наук. —1995. Т.35. С. 189—228.
13.
Сеpов В. В.// От
целлюляpной патологии Виpхова до молекуляpной патологии сегодняшнего дня, Архив
патологии №1, 2001, стр.3-6
14.
Скулачёв В. П.
Старение организма – особая биологическая функция, а не результат поломки
сложной биологической системы: биохимическое обоснование гипотезы Вейсмана
//Биохимия, 1997, том 62, вып. 11, с. 1394-1399
15.
Хейфлик Л. В кн.
Молекулы и клетки, Вып. 7, М., "Мир", 1982, 134-148.
16.
Хейфлик Л.
Смертность и бессмертие на клеточном уровне // Биохимия, 1997, том 62, вып. 11,
с. 1380-1393
17.
Baum
R. Superoxide theory of oxygen toxicity is center of heated debate. // Chem.
Engin. News.1984. V. 9. P. 20-28
18.
Brubaker
RF. Delayed functional loss in glaucoma. LII Edward Jackson Memorial Lecture.
Am J Ophthalmol 1996;121: 473-483
19.
Dreyer EB, Zurakowski D, Schumer RA, et al. Elevated glutamate
levels in the vitreous body of humans and monkeys with glaucoma. Arch
Ophthalmol 1996; 114:299-305
20.
Frade
JM, Bololenta P, Martinez-Morales JR, et al. Control of early cell death
by BDNF in chick retina. Development 1997;124:3313-3320
21.
Garcia-Valenzuela
E.; Shareef S.; Walsh J. Programmed cell death of retinal ganglion cells
during experimental glaucoma. Exp-Eye-Res. 1995 Jul; 61(1): 33-44
22.
Kerr F.R., Wyllie A.H., Currie A.R. Apoptosis: A basic biological
phenomenon with wide ranging implications in tissue kinetics. Br J Cancer 1972;
26:239-256
23.
Khan
A.U. //Science.1970. Vol.168. P.476-482. 1970
24.
Klocker N, Cellerino A, Bahr M. Free radical scavenging and
inhibition of nitric oxide synthase potentiates the neuro-trophic effects of
brain-derived neuro-trophic factor on axotomized retinal ganglion cells in
vivo. J Neurosci 1998;18:1038-1046
25.
Lash,
G. E., J. E. Cartwright, et al. (1999). “The effects of angiogenic growth
factors on extravillous trophoblast invasion and motility.” Placenta 20(8):
661-667
26.
Levin
L.A.; Louhab A. Apoptosis of retinal ganglion cells in anterior ischemic optic
neuropathy. Arch-Ophthalmol. 1996 Apr; 114(4): 488-91
27.
Lewen
A.; Matz P.; Chan P. J-Neurotrauma. 2000 17(10) P.871-90
28.
Li S.;
Chang C.J.; Abler A.S.; Fu J. A comparison of continuous versus intermittent
light exposure on apoptosis Curr-Eye-Res. 1996 Sep; 15(9): 914-22
29.
Macaya
A. Apoptosis in the nervous system Rev-Neurol. 1996 Nov; N24 Vol 135 P. 1356-60
30.
Miller
N.R.//Walsh and Hoyts clinical neuroophthalmology. 1982, Vol.1, P.175-176
31.
Minckler
D.S. Bunt A.H. Orthograde and retrograde axoplasmatic transport during acute
ocular hypertension in the monkey. Invest Ophthalm Vis Sci 1977 Vol.16, P.426
32.
Narayanan V. Apoptosis in development of the nervous system. 1.
Naturally occurring death in the developing nervous system. Pediatr Neurol 1997;
Vol 16 P. 9-13.
33.
Negoescu
A.; Guillermet C.; Lorimier P.// Biomed-Pharmacother. 1998; Vol 52 N6 P. 252-8
34.
Neufeld
A.H. Nitric oxide: a potential mediator of retinal ganglion cell damage in
glaucoma. Surv-Ophthalmol. 1999 Jun; 43 Suppl 1S129-35
35.
Nickells
R.W.; Zack D.J. Apoptosis in ocular disease: a molecular overview.
Ophthalmic-Genet. 1996 Dec; 17(4): 145-65
36.
Okisaka S, Murakami A, Mizukawa A . Apoptosis in retinal ganglion
cell decrease in human glaucoma eyes. Jpn J Ophthalmol 1997 Vol 41:84-88
37.
Ott, S.R., Jones, I.W.,
Burrows, M. and Elphick, M.R. () Sensory afferents and motor neurons as targets
for nitric oxide in the locust. J. Comp. Neurol. 2000 Vol. 422, P.521-532
38.
Pienardo-Ramon R, Salvador M., Villegas-Perez MP Effects of
axotomy and intraocular administration of NT-4, NT-3 and BDNF on the survival
of adult rat retinal ganglion cells: A quantitative in-vivo study. Inv
Ophthalmol Vis Sci 1996; 37:489-500
39.
Quigley
H.A. Ganglion cell death in glaucoma: pathology recapitulates ontogeny
Aust-N-Z-J-Ophthalmol. 1995 May; 23(2): 85-91
40.
Quigley
H.A.; Nickells R.W.; Kerrigan L.A Retinal ganglion cell death in experimental
glaucoma and after axotomy occurs by apoptosis. . Invest-Ophthalmol-Vis-Sci. 1995 Apr; 36(5): 774-86
41.
Rait
J. The prevention of ganglion cell
suicide in primary open angle glaucoma Asian J Ophthalmol
1998;1(2):3-8
42.
Roschupkin
D.I., Pelenitsin A.B., Potapenko A. // Photochem.and Photobiol. 1975.
Vol.21.P.63-69
43.
Saunders,
J.W. 1966. Death in Embryonic Systems. Science 154: 604-612.
44.
Schapira,-A-H
Mitochondrial disorders. Curr-Opin-Neurol. 1997 Feb; 10(1): 43-7
45.
Schwartz
M., Belkin M., Yoles E. Potential treatment modalities for glaucomatous
neuropathy: neuroprotection and neuroregeneration. J Glaucoma 1996;5:427-432
46.
Sieving
P. A. Electrical Signals of the Retina and Visual Cortex. Digital Duane’s
Ophthalmology Vol. 2, Chapter 13
47.
Snyder,
Solomon H., "Nitric Oxide: First in a New Class of
Neurotransmitters?", Science, Vol. 257, 1992, P. 494-496.
48.
Steller,
H. 1995. Mechanisms and genes of cellular suicide. Science 267: 1445-1449
49.
Sun D.
Y., Jiang S., Zheng L.-M., et. al. (1995) J. Exp. Med., 179, 559-568
50.
Wadia J.S., Chalmers-Redman R., Ju W., et al. Mitochondrial
membrane potential and nuclear changes in apoptosis caused by serum and nerve
growth factor withdrawal: time course and modification by (-) -Deprenyl. J
Neurosci 1998;18:932-947